Admixture Mapping¶
Admixture mapping tests whether local ancestry at a genomic region is associated with a phenotype in recently admixed individuals. Instead of asking whether a specific SNP allele is associated with a trait, we ask whether carrying ancestry from a source population at a local genomic window changes trait values after accounting for background ancestry and other covariates.
In this tutorial, we build a synthetic local ancestry dataset with a quantitative trait, run snputils.tools.run_admixture_mapping, and make a Manhattan plot from the resulting association table.
from pathlib import Path
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
from snputils.datasets import build_synthetic_admixture_dataset
from snputils.phenotype.genobj import CovariateObject, PhenotypeObject
from snputils.tools import run_admixture_mapping
from snputils.visualization import manhattan_plot, qq_plot
What the input represents¶
A local ancestry inference method usually produces one ancestry label per haplotype per genomic window. For a diploid sample, each window therefore has two ancestry labels: one for each haplotype. snputils stores that information in a LocalAncestryObject.
For each ancestry and each window, admixture mapping converts those two haplotype labels into an ancestry dosage: 0, 1, or 2 copies of the requested ancestry. The association model then tests whether that dosage is related to the phenotype.
Build a synthetic dataset¶
The synthetic trait below has a few sparse local ancestry effects plus global ancestry-like covariates, so the strongest signals should appear near the simulated causal windows while most tests remain null.
build_synthetic_admixture_dataset lives in snputils.datasets, and returns:
laiobj: local ancestry calls for each haplotype and window.phenotype: a continuous trait generated from a few local ancestry effects, covariates, and noise.covar_matrix: covariates that include genome-wide ancestry-like summaries.effect_windows,effect_ancestries, andeffect_sizes: the simulated ground truth used only for interpretation.
RESULTS_DIR = Path("results/admixture_mapping")
RESULTS_DIR.mkdir(parents=True, exist_ok=True)
PHENO_NAME = "TRAIT"
synthetic = build_synthetic_admixture_dataset(
n_samples=2000,
n_windows=22000,
n_covariates=3,
seed=42,
)
laiobj = synthetic["laiobj"]
sample_ids = synthetic["sample_ids"]
phenotype_values = synthetic["phenotype"]
covar_names = synthetic["covar_names"]
covar_matrix = synthetic["covar_matrix"]
print(laiobj)
print(f"Phenotype shape: {phenotype_values.shape}")
print(f"Covariate matrix shape: {covar_matrix.shape}")
LocalAncestryObject(shape=(22000, 4000), n_windows=22000, n_samples=2000, n_haplotypes=4000, n_ancestries=3, has_window_metadata=True, has_ancestry_map=True)
Phenotype shape: (2000,)
Covariate matrix shape: (2000, 3)
The simulated causal windows are shown below. In real data these columns do not exist; they are included here so we can sanity-check whether the analysis recovers the planted signal.
truth = pd.DataFrame({
"window_index_0_based": synthetic["effect_windows"],
"result_window_index_1_based": synthetic["effect_windows"] + 1,
"chromosome": laiobj.chromosomes[synthetic["effect_windows"]],
"start": laiobj.physical_pos[synthetic["effect_windows"], 0],
"end": laiobj.physical_pos[synthetic["effect_windows"], 1],
"ancestry_code": synthetic["effect_ancestries"],
"ancestry": [laiobj.ancestry_map[str(code)] for code in synthetic["effect_ancestries"]],
"effect_size": synthetic["effect_sizes"],
})
truth["expected_result_id"] = [
f"w{window_idx}_{ancestry}"
for window_idx, ancestry in zip(truth["result_window_index_1_based"], truth["ancestry"])
]
truth
| window_index_0_based | result_window_index_1_based | chromosome | start | end | ancestry_code | ancestry | effect_size | expected_result_id | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 2200 | 2201 | 3 | 5000001 | 5004999 | 0 | AFR | 0.30 | w2201_AFR |
| 1 | 12099 | 12100 | 13 | 2475001 | 2479999 | 1 | EUR | -0.26 | w12100_EUR |
| 2 | 21999 | 22000 | 22 | 24975001 | 24979999 | 2 | AMR | 0.22 | w22000_AMR |
Create phenotype and covariate inputs¶
run_admixture_mapping accepts file paths or in-memory snputils objects. Here we use an in-memory PhenotypeObject, LocalAncestryObject, and CovariateObject.
Covariates are important in real admixture mapping. Local ancestry is correlated across the genome and with global ancestry; without adjustment, a local window can look associated because it tags broader ancestry structure rather than a region-specific effect.
phenotype = PhenotypeObject(
samples=sample_ids,
values=phenotype_values,
phenotype_name=PHENO_NAME,
quantitative=True,
)
covariates = CovariateObject(
samples=sample_ids,
values=covar_matrix,
covariate_names=covar_names,
)
pd.DataFrame(covariates.values, index=covariates.samples, columns=covariates.covariate_names)
| COV1 | COV2 | COV3 | |
|---|---|---|---|
| A00000 | -0.330546 | 0.634283 | 0.236212 |
| A00001 | -0.651705 | 1.062672 | 0.336365 |
| A00002 | -0.275401 | -0.115268 | -0.084457 |
| A00003 | -0.753692 | 1.544846 | 0.866777 |
| A00004 | 0.401226 | 0.367596 | -0.915346 |
| ... | ... | ... | ... |
| A01995 | -0.921008 | 0.516885 | 0.657766 |
| A01996 | -1.158979 | 2.452996 | 2.187145 |
| A01997 | 1.127358 | -1.241836 | 0.632129 |
| A01998 | -0.219785 | -0.030625 | 0.777080 |
| A01999 | 0.640294 | 0.621354 | 0.687112 |
2000 rows × 3 columns
Run admixture mapping¶
The API uses lai_source for either an MSP path or a LocalAncestryObject.
For a quantitative trait, snputils fits a linear model per window and ancestry. For a binary trait, it fits a logistic model. The returned table includes p-values, effect estimates, standard errors, test type, ancestry labels, chromosome, position, and multiple-testing adjusted p-values when adjust=True.
results_path = RESULTS_DIR / "synthetic_admixture_mapping.tsv.gz"
results = run_admixture_mapping(
phe_path=phenotype,
lai_source=laiobj,
results_path=results_path,
batch_size=256,
quantitative=True,
covar=covariates,
covar_variance_standardize=True,
ci=0.95,
adjust=True,
keep_hla=True,
return_results=True,
verbose=True,
)
results
Reading LAI source (LocalAncestryObject)...
Chunk 0: 256 windows (total so far: 256)
Done. Processed 22,000 windows.
| #CHROM | POS | END | ID | REF | ALT | A1 | ANCESTRY | TEST | OBS_CT | BETA | SE | T_STAT | P | L95 | U95 | BONF | FDR_BH | ERRCODE | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 1 | 4999 | w1_AFR | N | AFR | AFR | AFR | LINEAR | 2000 | -0.171130 | 0.087126 | -1.964177 | 4.964817e-02 | -0.341997 | -0.000263 | 1.000000 | 0.949899 | . |
| 1 | 1 | 25001 | 29999 | w2_AFR | N | AFR | AFR | AFR | LINEAR | 2000 | -0.214909 | 0.086942 | -2.471884 | 1.352322e-02 | -0.385415 | -0.044404 | 1.000000 | 0.869918 | . |
| 2 | 1 | 50001 | 54999 | w3_AFR | N | AFR | AFR | AFR | LINEAR | 2000 | -0.223219 | 0.086726 | -2.573841 | 1.012925e-02 | -0.393302 | -0.053136 | 1.000000 | 0.852834 | . |
| 3 | 1 | 75001 | 79999 | w4_AFR | N | AFR | AFR | AFR | LINEAR | 2000 | -0.237153 | 0.086625 | -2.737707 | 6.241845e-03 | -0.407038 | -0.067269 | 1.000000 | 0.761482 | . |
| 4 | 1 | 100001 | 104999 | w5_AFR | N | AFR | AFR | AFR | LINEAR | 2000 | -0.233500 | 0.086671 | -2.694094 | 7.117167e-03 | -0.403476 | -0.063525 | 1.000000 | 0.792420 | . |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 65995 | 22 | 24875001 | 24879999 | w21996_AMR | N | AMR | AMR | AMR | LINEAR | 2000 | 0.442430 | 0.098918 | 4.472673 | 8.159219e-06 | 0.248436 | 0.636424 | 0.538508 | 0.024478 | . |
| 65996 | 22 | 24900001 | 24904999 | w21997_AMR | N | AMR | AMR | AMR | LINEAR | 2000 | 0.490311 | 0.100017 | 4.902289 | 1.023791e-06 | 0.294163 | 0.686459 | 0.067570 | 0.006757 | . |
| 65997 | 22 | 24925001 | 24929999 | w21998_AMR | N | AMR | AMR | AMR | LINEAR | 2000 | 0.483034 | 0.099555 | 4.851932 | 1.317446e-06 | 0.287791 | 0.678277 | 0.086951 | 0.007723 | . |
| 65998 | 22 | 24950001 | 24954999 | w21999_AMR | N | AMR | AMR | AMR | LINEAR | 2000 | 0.509389 | 0.100400 | 5.073602 | 4.265379e-07 | 0.312489 | 0.706288 | 0.028152 | 0.004685 | . |
| 65999 | 22 | 24975001 | 24979999 | w22000_AMR | N | AMR | AMR | AMR | LINEAR | 2000 | 0.534291 | 0.099897 | 5.348402 | 9.891250e-08 | 0.338377 | 0.730205 | 0.006528 | 0.003264 | . |
66000 rows × 19 columns
Inspect the result table¶
Each row is one ancestry-specific test at one local ancestry window. P is the nominal association p-value. BETA is the estimated change in the quantitative trait per additional copy of that local ancestry, after covariate adjustment.
result_cols = [
"#CHROM", "POS", "ID", "ANCESTRY", "TEST", "OBS_CT", "BETA", "SE", "P", "ERRCODE"
]
optional_cols = ["L95", "U95", "BONF", "FDR_BH"]
show_cols = [col for col in result_cols + optional_cols if col in results.columns]
results[show_cols].sort_values("P").head(10)
| #CHROM | POS | ID | ANCESTRY | TEST | OBS_CT | BETA | SE | P | ERRCODE | L95 | U95 | BONF | FDR_BH | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
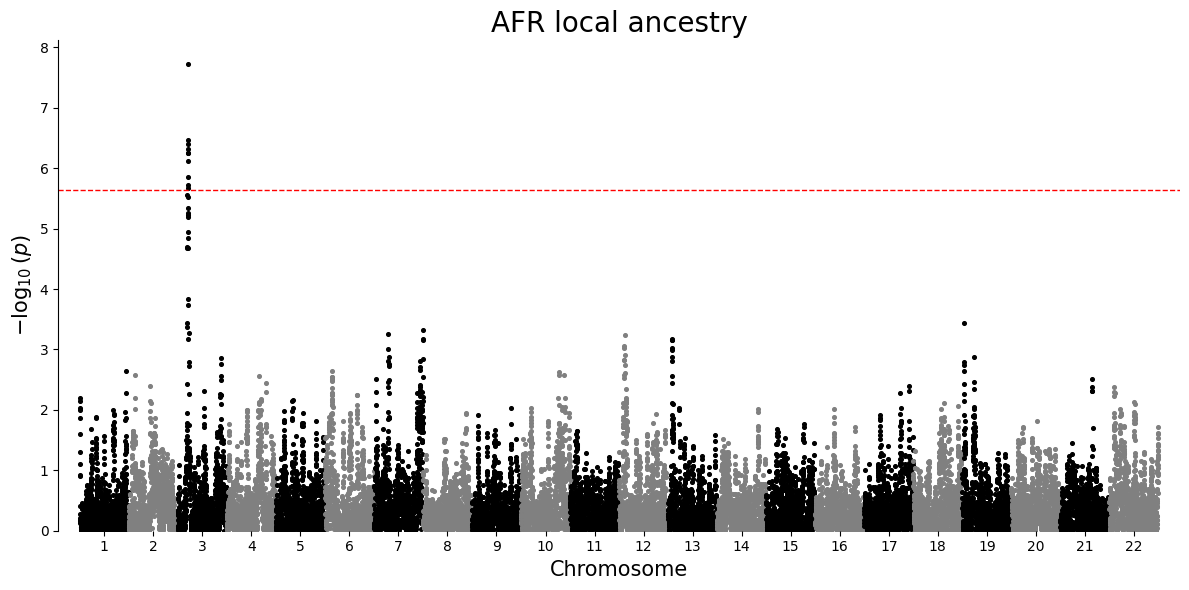
| 6296 | 3 | 5000001 | w2201_AFR | AFR | LINEAR | 2000 | 0.464202 | 0.082196 | 1.861255e-08 | . | 0.303003 | 0.625400 | 0.001228 | 0.001228 |
| 65999 | 22 | 24975001 | w22000_AMR | AMR | LINEAR | 2000 | 0.534291 | 0.099897 | 9.891250e-08 | . | 0.338377 | 0.730205 | 0.006528 | 0.003264 |
| 6299 | 3 | 5075001 | w2204_AFR | AFR | LINEAR | 2000 | 0.423219 | 0.082736 | 3.432297e-07 | . | 0.260962 | 0.585477 | 0.022653 | 0.004685 |
| 6297 | 3 | 5025001 | w2202_AFR | AFR | LINEAR | 2000 | 0.420844 | 0.082709 | 3.952853e-07 | . | 0.258639 | 0.583049 | 0.026089 | 0.004685 |
| 65998 | 22 | 24950001 | w21999_AMR | AMR | LINEAR | 2000 | 0.509389 | 0.100400 | 4.265379e-07 | . | 0.312489 | 0.706288 | 0.028152 | 0.004685 |
| 6305 | 3 | 5225001 | w2210_AFR | AFR | LINEAR | 2000 | 0.422560 | 0.083664 | 4.803442e-07 | . | 0.258482 | 0.586638 | 0.031703 | 0.004685 |
| 6295 | 3 | 4975001 | w2200_AFR | AFR | LINEAR | 2000 | 0.414107 | 0.082419 | 5.499524e-07 | . | 0.252471 | 0.575743 | 0.036297 | 0.004685 |
| 6302 | 3 | 5150001 | w2207_AFR | AFR | LINEAR | 2000 | 0.416082 | 0.082915 | 5.678927e-07 | . | 0.253473 | 0.578690 | 0.037481 | 0.004685 |
| 6306 | 3 | 5250001 | w2211_AFR | AFR | LINEAR | 2000 | 0.414821 | 0.083673 | 7.738177e-07 | . | 0.250726 | 0.578917 | 0.051072 | 0.005675 |
| 65996 | 22 | 24900001 | w21997_AMR | AMR | LINEAR | 2000 | 0.490311 | 0.100017 | 1.023791e-06 | . | 0.294163 | 0.686459 | 0.067570 | 0.006757 |
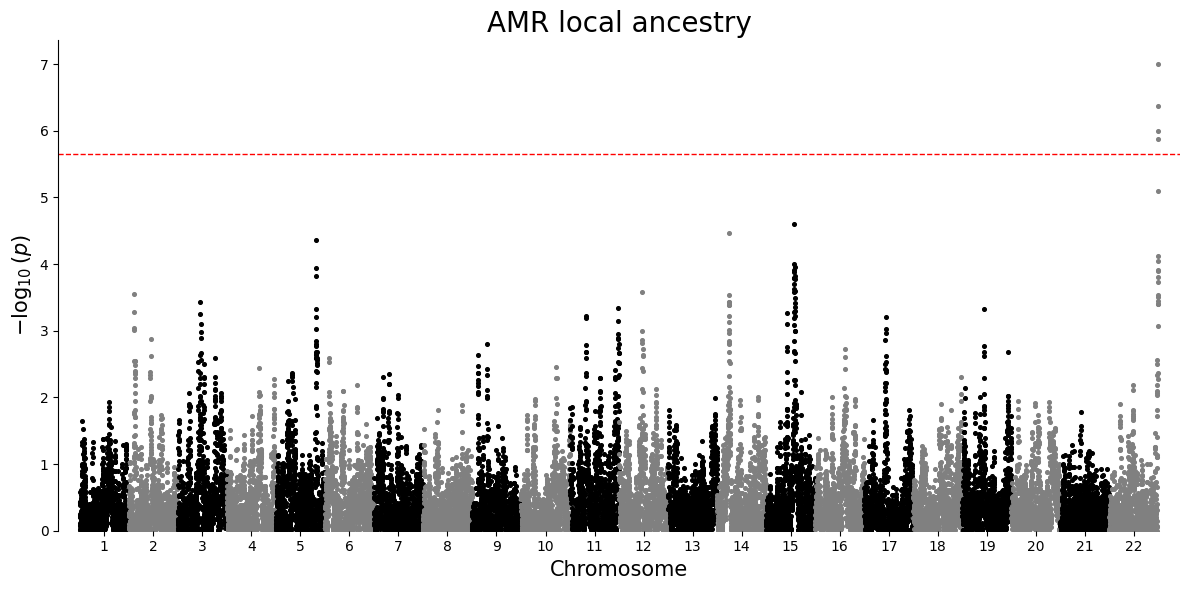
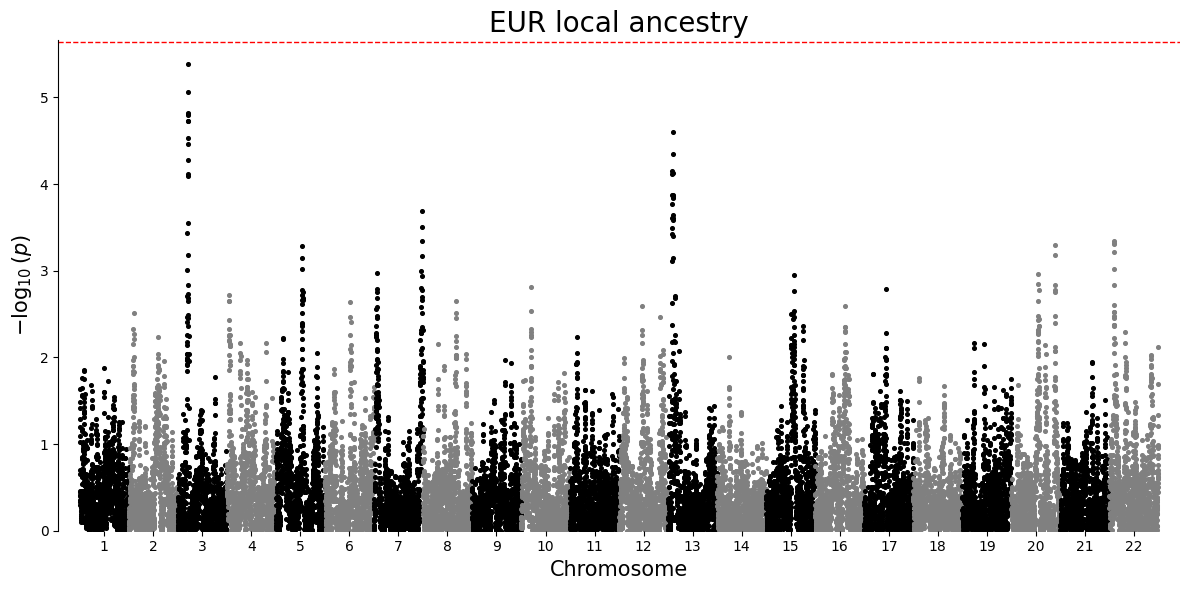
Manhattan plot¶
A Manhattan plot places association strength along genomic position. Each point is one local ancestry window for a single ancestry-specific test. Taller points have smaller p-values. The dashed line is a Bonferroni threshold, a strict correction that divides the chosen significance level by the number of tests in that ancestry-specific plot.
plot_df = results.loc[results["P"].notna() & (results["P"] > 0), ["#CHROM", "POS", "P", "ANCESTRY"]].copy()
for ancestry in sorted(plot_df["ANCESTRY"].unique()):
ancestry_df = plot_df.loc[plot_df["ANCESTRY"] == ancestry, ["#CHROM", "POS", "P"]].copy()
manhattan_plot(ancestry_df, title=f"{ancestry} local ancestry")
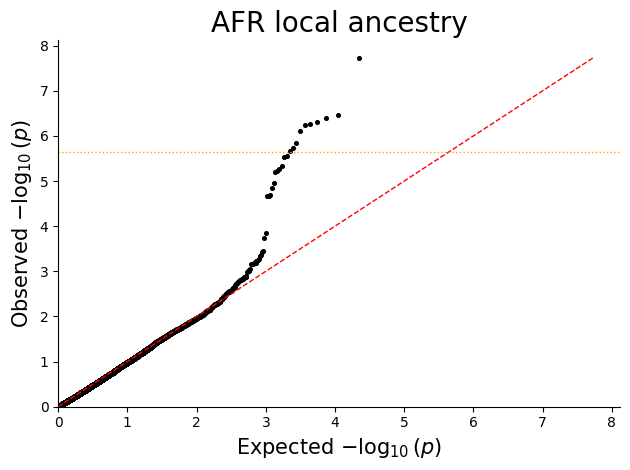
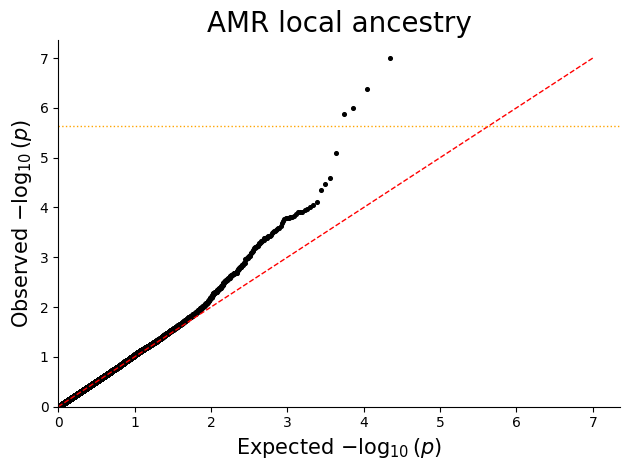
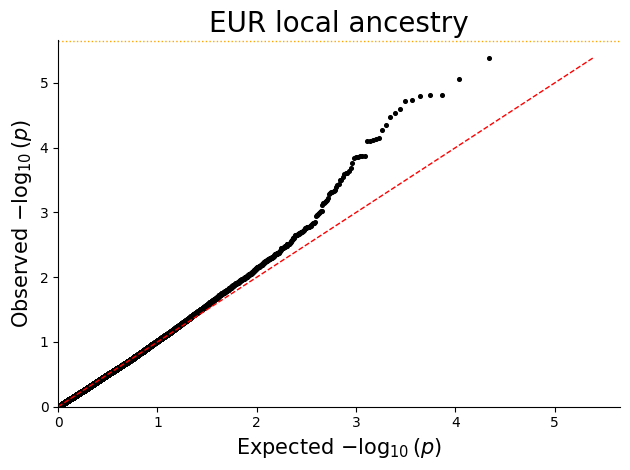
Q-Q plot¶
Q-Q plots help check whether the p-value distribution looks well calibrated under the null and highlight departures consistent with true signal or residual confounding.
for ancestry in sorted(plot_df["ANCESTRY"].unique()):
ancestry_df = plot_df.loc[plot_df["ANCESTRY"] == ancestry, ["P"]].copy()
qq_plot(ancestry_df, title=f"{ancestry} local ancestry")